Resolving hidden states of flexible biomolecules with iterative structure refinements
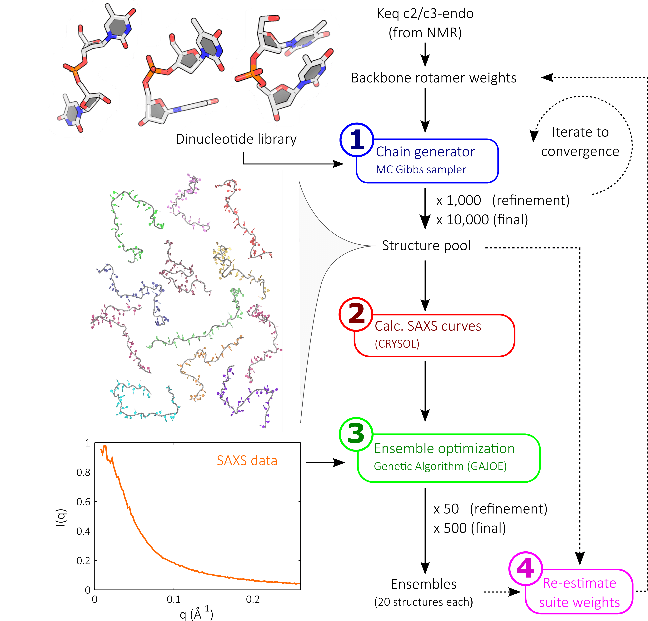
Biomolecules exist in multiple different conformations, constantly cajoled and buffeted by water, ions, and biological partners, forcing their structures to adapt in response. Many of these perturbing forces so overwhelm the biomolecule that thousands of conformations are visited every millisecond. Information on these states is hard to access, as traditional techniques probe only the average conformation, hiding all the fluctuations. By pairing solution X-ray scattering with advanced structure building methods, we gain access to these previously invisible states. These states often hide key centers for biological reactions such as catalysis and binding.
Our structure building method (illustrated right [1]) consists of constructing nucleic acids step-by-step, one base at a time, drawing probabilistically from a library of possible building blocks. Repeating this procedure allows us to build a large pool of possible biomolecule states. By comparing these possible states to solution X-ray scattering data, those which most closely resemble the experiment are identified. Based on the steps present in these selected structures, the library of building blocks is updated and reweighted. More structures are built from this updated pool, and the process iterated until convergence.
When the dust settles, we are left with sets of states reflecting all possible conformations of our biomolecule, as shown in the panel below. This provides an unprecedented view of molecular conformations [2].

Computer modeling of DNA-histone interactions

In the Pollack lab, we are working to better understand DNA-protein interactions using computer modeling. Our lab’s favorite technique, small angle x-ray scattering (SAXS), does well for systems with discrete states (like many proteins and RNA); however, understanding and interpreting the results of continuous systems is much more challenging. This makes looking at changes in DNA structure with SAXS a very interesting problem that computational modeling approaches are well suited for.
A system of particular interest to us is the interaction between DNA and histone proteins. Four protein varieties combine to form an octamer core, which wraps DNA around itself in two turns to form a nucleosomal complex. These nucleosomes are the fundamental organization and storage system of DNA in cells and interact to form more complex storage structure. In order to understand this interaction, we use SAXS to observe the unwrapping of the DNA for various modifications of the DNA and protein core.

In order to model this, we start with the structure of the wrapped DNA. We then use model all of the inter- and intra-base pair motions as having spring potentials based on which bases present. We can then allow certain portions of the nucleosome to move and look for their natural equilibrium. From this equilibrium we can also look for fluctuations that have energies that we would expect from a solution at room temperature. This process allows us to quickly generate tens of thousands of unwrapping structures, without needing to know anything at all about the protein-DNA interactions ahead of time. By simulating the scattering off each of these structures, we can find representative groups that fit our data, which gives us insight into how the unwrapping progresses.
References
- Plumridge et al. Visualizing single-stranded nucleic acids in solution, Nucleic Acids Research, 2016. doi: 10.1093/nar/gkw1297
- Plumridge et al. The impact of base stacking on the conformations and electrostatics of single-stranded DNA. Nucleic Acids Research, 2017. doi: 10.1093/nar/gkx140